ФІЗИКА ДЛЯ БАКАЛАВРІВ. МОЛЕКУЛЯРНА ФІЗИКА ТА ТЕРМОДИНАМІКА
VIII. ЕНТРОПІЯ
Теорема Карно не тільки визначає обмеження, накладені природою на можливість перетворення теплової енергії на механічну. З неї випливає існування фізичної величини, що дозволє аналітично виразити друге начало термодинаміки й висвітлює його фундаментальний зміст. Ця величина, котра називається ентропією, розглядається в наступних питаннях:
2. Обчислення ентропії. Теорема Нернста
3. Нерівність Клаузіуса. Закон зростання ентропії
4. Ентропія й імовірність. Формула Больцмана
\({1+\frac{Q_2}{Q_1}}=1-\frac{T_2}{T_1}\) \(\Rightarrow \) \(\frac{Q_2}{Q_1}=-\frac{T_2}{T_1}\) \(\Rightarrow \) \(\frac{Q_1}{T_1}=\frac{Q_2}{T_2}\),
або
|
\(\frac{Q_1}{T_1}+\frac{Q_2}{T_2}=0 \). |
(8.1) |
|
|
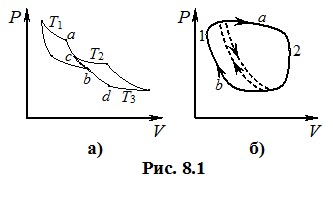
Клаузіус назвав відношення кількості теплоти, отриманої системою, до її температури зведеною кількістю теплоти. Тож сума зведених кількостей теплоти, отриманих системою в циклі Карно, дорівнює нулю. Відтак розглянемо оборотний коловий процес, який складається з чотирьох ізотерм і чотирьох адіабат (рис. 8.1а), ділянки а-b і с-d яких лежать на одній адіабаті (точки b і с не з’єднані). Якщо подумки продовжити адіабату ас від точки с до точки b, а адіабату db – від точки b до точки с, то вказаний комбінований процес розпадається на два окремі цикли Карно, котрі здійснюються між температурами Т1 і Т4 та Т2 і Т3. При цьому, позаяк вставлені віртуальні процеси між точками b і с є адіабатичними (Q = 0), то вони ніяк не впливають на зведені теплоти. Відтак для всього “ступінчастого” циклу маємо:
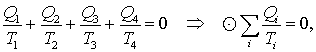 |
(8.2) |
де кружечок біля знаку суми означає, що додавання виконується по всьому коловому процесу (по замкненому “шляху”). Довільний коловий процес (рис. 8.1б) також можна розчленувати віртуальними адіабатами на безліч елементарних циклів Карно (на рис.8.1б штрихами показано один із таких елементарних циклів). При цьому на кожній елементарній ділянці циклу система отримує нескінченно малу зведену кількість теплоти \(\delta{Q}/{T}\), тому дискретна сума (8.2) вироджується в інтеграл по всьому коловому процесу:
|
\(\oint\frac{\delta{Q}}{T}=0 \). |
(8.3) |
Вирази (8.1), (8.2) і (8.3) називаються рівностями Клаузіуса. Вони означають, що
в будь-якому оборотному коловому процесі сумарна зведена кількість теплоти дорівнює нулю.
Зауважимо, що найбільш загальним є вираз (8.3), а (8.1) і (8.2) є його окремими випадками.
Ентропія. Отриманий результат можна подати інакше. Поділимо весь цикл (рис. 8.1 б) на дві ділянки: ![]() та
та  і, відповідно, інтеграл (8.3), на дві частини. Тоді
і, відповідно, інтеграл (8.3), на дві частини. Тоді
|
\(\oint\frac{\delta{Q}}{T}=\int_{1a2}\frac{\delta{Q}}{T}+\int_{2b1}\frac{\delta{Q}}{T}=0 \quad\Rightarrow \quad \int_{1a2}\frac{\delta{Q}}{T}=-\int_{2b1}\frac{\delta{Q}}{T}\). |
(8.3а) |
Оскільки цикл оборотний, то при проведенні процесу  у зворотному напрямку по шляху
у зворотному напрямку по шляху  в кожній точці зміниться тільки напрям теплообміну й знак величини \(\delta{Q}/{T}\) , тому
в кожній точці зміниться тільки напрям теплообміну й знак величини \(\delta{Q}/{T}\) , тому
\({-}\int_{2b1}\frac{\delta{Q}}{T}=\int_{1b2}\frac{\delta{Q}}{T}\)
Отже, (8.3а) можна переписати, як
|
\(\int_{1a2}\frac{\delta{Q}}{T}=\int_{1b2}\frac{\delta{Q}}{T}\). |
(8.4) |
Це дуже важливий результат, який означає, що при оборотному переході системи із якогось стану 1 у якийсь стан 2 сумарна зведена кількість теплоти, отримана системою, не залежить від способу переходу (тобто, виду процесу) й визначається тільки параметрами початкового та кінцевого стану системи. Схожа ситуація спостерігається в механіці[1] стосовно роботи консервативних сил, причому незалежність вказаної роботи від траєкторії руху частинки дозволяє ввести поняття потенціальної енергії. Подібним чином у термодинаміці вводиться поняття ентропії S.
Ентропією системи називається величина, зміна котрої при переході системи з одного стану в інший будь-яким квазістатичним (оборотним) способом дорівнює сумарній зведеній кількості теплоти, що її отримує система при цьому переході:
|
\(\Delta{S}=S_2 - S_1=\left(\int_{1\to{2}}\frac{\delta{Q}}{T}\right) \)об. |
(8.5) |
Відповідно, при елементарній зміні стану приріст ентропії
|
\(\mathrm{d}S=\frac{\delta{Q}}{T}\). |
(8.5а) |
Одразу звернімо увагу на такі загальні властивості ентропії. По-перше, хоча кількість теплоти є функцією процесу, величина \(\mathrm{d}S=\delta{Q}/{T}\) і, відповідно, ΔS не залежить від способу зміни стану, отже,
ентропія є функцією стану системи.
По-друге, кількість теплоти, отримана системою в будь-якому квазістатичному процесі, є прямо пропорційною масі (кількості речовини) системи. Тому
ентропія є адитивною величиною,
тобто, ентропія системи дорівнює сумі ентропій її окремих частин. Це дозволяє поширити поняття ентропії на нерівноважні стани системи, якщо її можна поділити на окремі частини (підсистеми), що перебувають у відмінних, але квазірівноважних станах. Нарешті, з (8.3) і (8.5) випливає що в оборотному коловому процесі
|
\(\Delta{S}={0}\), |
(8.6) |
тобто,
в оборотних колових процесах ентропія не змінюється.
Ентропія ідеального газу. Нехай ідеальний газ у кількості ν молів оборотно переходить із стану 1 (P1, V1, T1) у стан 2 (P2, V2, T2). Визначимо зміну ентропії газу. Згідно з першим началом термодинаміки (4.14), при елементарній зміні стану системи \(\delta{Q}=\mathrm{d}U+P\mathrm{d}{V}\). Урахувавши (5.7) і (2.6), будемо мати: \(\delta{Q}=\nu{C_V}\mathrm{d}T+\nu{RT}\mathrm{d}V/{V}\). Звідси для елементарної зведеної кількості теплоти дістанемо:
|
\(\frac{\delta{Q}}{T}=\nu{C_V}\cdot\frac{\mathrm{d}T}{T}+\nu{R}\cdot\frac{\mathrm{d}V}{V}\) |
(8.7) |
Інтегруючи цей вираз відповідно до (8.5), отримаємо
\(\Delta{S}=\nu{C_V}\int_{T_1}^{T_2} \frac{\mathrm{d}T}{T}+\nu{R}\int_{V_1}^{V_2} \frac{\mathrm{d}V}{V}\quad\Rightarrow\)
|
\(\Rightarrow\quad\Delta{S}=S_2-S_1=\nu\left(C_V\ln\frac{T_2}{T_1}+R\ln\frac{V_2}{V_1}\right) \). |
(8.8) |
Величину ΔS можна подати й через інші параметри стану, зробивши у (8.8) відповідні заміни на основі рівняння Клапейрона (2.6). Зокрема, можна отримати такий симетричний вираз:
|
\(\Delta{S}=\nu\left(C_V\ln\frac{P_2}{P_1}+C_P\ln\frac{V_2}{V_1}\right)\). |
(8.8а) |
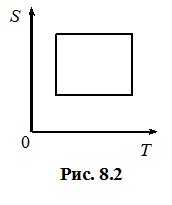
З (8.8) і (8.8а) випливають прості формули для обчислення зміни ентропії ідеального газу в ізопроцесах. До прикладу, в ізотермічному процесі \(\Delta{S}=\nu{R}\ln (V_2/V_1)\), або \(\Delta{S}=\nu{R}\ln{(P_1/P_2)}\), в ізобарному процесі \(\Delta{S}=\nu{C_P}\ln{(V_2/V_1)}\), або \(\Delta{S}=\nu{C_P}\ln{(T_1/T_1)}\) в ізохорному процесі \(\Delta{S}=\nu{C_V}\ln{(P_2/P_1)}\), або \(\Delta{S}=\nu{C_V}\ln{(T_2/T_1)}\). Принагідно відмітимо, що в адіабатному процесі \(\delta{Q}={0}\), тому й ΔS = 0. Отже, адіабатний процес – то є ізоентропний (ізоентропійний) процес. Відповідно, на діаграмі (T, S) графік циклу Карно зображується прямокутником (рис. 8.2).
 |
Це, зокрема, дозволяє дуже легко отримати вираз (7.4) для ККД циклу Карно. Справді, оскільки в цьому циклі теплообмін відбувається тільки на ізотермах при сталих температурах T1 і T2, то, відповідно до (8.5), Q1 = T1(S2 - S1) і Q2 = T2(S2 - S1). Підставивши ці вирази в (7.2) з урахуванням знаків, одразу отримуємо формулу (7.4).
Зміна ентропії при необоротній зміні стану газу. Нехай в одній із двох теплоізольованих посудин, які з’єднані тонкою трубкою з перекритим краном, знаходиться ν молів ідеального газу, а в другій – вакуум. Об’єми посудин V1 і V2. Кран відкривають і газ, розширюючись в пустоту, заповнює обидві посудини й переходить у новий рівноважний стан. Такий процес є необоротним: для повернення газу у вихідний стан його треба стиснути, а для цього треба виконати роботу та забезпечити теплообмін, що суттєво змінить стан зовнішніх тіл. Тому годі навіть і думати про обчислення зміни ентропії за допомогою виразу (8.5) безпосередньо для цього процесу, адже для необоротного процесу саме поняття зведеної кількості теплоти втрачає зміст через невизначеність температури системи. Але розширення ідеального газу в пустоту відбувається без зміни температури[2], тому замість реального процесу можна розглянути квазістатичне ізотермічне розширення газу від об’єму V1 до об’єму V1 + V2. Тоді, з урахуванням (4.15) і (2.8), із (8.5) отримаємо:
|
\(\Delta{S}=\int_{V_1}^{V_1+V_2}\frac{P\mathrm{d}V}{T}=\nu{R}\int_{V_1}^{V_1+V_2}\frac{\mathrm{d}V}{V}\quad\Rightarrow\quad\Delta{S}=\nu{R}\ln\frac{V_1+V_2}{V_1}\) |
(8.9) |
Отже, в розглянутому процесі ентропія газу зростає.
Теорема Нернста. Означення (8.5) визначає не саму ентропію, а тільки її зміну при заданій зміні стану системи. Тому для визначення самої ентропії необхідно в будь-якому обраному стані покласти ентропію рівною нулю, подібно до того, як це робиться в механіці стосовно потенціальної енергії[2]. Але у випадку ентропії вибір нульового рівня, по суті, є однозначним, завдяки так званій тепловій теоремі Нернста. Нернст на основі емпіричних даних установив, що при наближенні температури до 0 К ентропія будь-якої системи прямує до певної границі, що не залежить від жодних параметрів стану системи. Тому цю границю покладають рівною нулю, і приймають таке формулювання:
при наближенні температури до абсолютного нуля ентропія будь-якої системи прямує до нуля:
|
\({S}\to{0}\), якщо \({T}\to {0}\). |
(8.10) |
Слід зауважити, що теорема Нернста логічно не випливає з інших законів термодинаміки. Тому її ще називають третім началом термодинаміки.
Теорема Нернста дозволяє дати означення не лише для зміни, а й для самої ентропії системи:
|
\({S}=\int_0^T\frac{\delta{Q}}{T}\). |
(8.11) |
|
\( \frac{Q_1}{T_1}+\frac{Q_2}{T_2}\le{0}\), |
(8.12) |
|
|
|
(8.13) |
|
|
\(\oint\frac{\delta{Q}}{T}\le{0}\), |
(8.14) |
в яких знак “<” відноситься до необоротних, а знак “=”– до оборотних колових процесів.
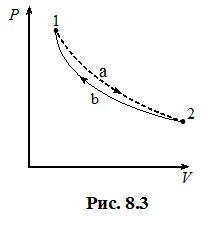 |
Нерівність (8.14) дозволяє з’ясувати загальну поведінку ентропії в необоротних процесах. Нехай система перейшла зі стану 1 у стан 2 в якомусь необоротному процесі, показаному умовно[3] штриховою лінією  на рис. 8.3. Повернемо подумки систему у вихідний стан, здійснивши певний оборотний процес, який показано лінією . Відтак отримуємо необоротний цикл, для якого, згідно з (8.14) можна записати:
на рис. 8.3. Повернемо подумки систему у вихідний стан, здійснивши певний оборотний процес, який показано лінією . Відтак отримуємо необоротний цикл, для якого, згідно з (8.14) можна записати:
\(\int_{1a2}\frac{\delta{Q}}{T}+\int_{2b1}\frac{\delta{Q}}{T}\lt{0}\).
У першому інтегралі під Т слід розуміти температуру навколишнього середовища, при якій відбувається теплообмін, у другому інтегралі ця температура є й температурою системи. Процес є оборотним, тому, згідно з (8.5), другий інтеграл дорівнює зміні ентропії S1 – S2 при переході системи із стану 2 у стан 1. Отже,
|
\(\int_{1a2}\frac{\delta{Q}}{T}+(S_1-S_2)\lt{0}\quad\rightarrow\quad{S_2-S_1}\gt\int_{1a2}\frac{\delta{Q}}{T}\). |
(8.15) |
Зведена кількість теплоти (інтеграл у правій частині кінцевої нерівності), яку отримує система, може мати будь-який знак, залежно від напрямку та інтенсивності теплообміну на окремих стадіях процесу. Тому в довільному необоротному процесі ентропія системи може як зростати, так і зменшуватись. Але, якщо система є теплоізольованою, тобто, не обмінюється теплом із навколишніми тілами, то інтеграл у (8.15) дорівнює нулю, відтак ΔS = S2 – S1 > 0. Отже, при необоротних процесах у теплоізольваній системі ентропія обов’язково зростає. Об’єднуючи цей результат із (8.6), отримуємо універсальне співвідношення
|
\(\Delta{S}\ge{0}\), |
(8.16) |
яке означає, що
ентропія теплоізольованої системи не може зменшуватися – вона або зростає, або лишається незмінною.
Цей результат називають законом зростання ентропії. Він безпосередньо випливає з теореми Карно, котра, у свою чергу, є прямим наслідком другого начала термодинаміки і, по суті, виражає його. Тому закон зростання ентропії (8.16) розглядають як ще одне формулювання другого начала термодинаміки. З нього, зокрема, випливає висновок про те, що в замкнених системах можливі лише такі процеси, при яких ентропія не зменшується. А практично,позаяк строго оборотних процесів у природі не існує, в усіх процесах у замкнених системах ентропія зростає[4]. Тим самим, друге начало термодинаміки визначає напрям процесів, які можуть відбуватися в природі.
4.Ентропія й імовірність. Формула Больцмана
умовою термодинамічної рівноваги системи є максимум її ентропії.
В цьому сенсі можна говорити про те, що ентропія показує ступінь наближеності системи до стану рівноваги: чим ближча ентропія до свого максимуму, тим ближчим є стан системи до рівноважного. Така думка підтримується й тим, що перехід замкненої системи до рівноваги є необоротним, а саме необоротність термодинамічних процесів є формальною причиною зростання ентропії у замкнених системах. Тому постає питання про причину цієї необоротності, адже термодинамічні процеси визначаються рухом і взаємодією між частинками макроскопічної системи, а в законах руху (законах механіки) ніякої необоротності немає. Наприклад, якби Земля рухалася навколо Сонця в протилежному до існуючого напрямку, ні на Землі, ні у Всесвіті взагалі, нічого б не змінилось.
Виявляється, що причина необоротності теплових процесів полягає в макроскопічності термодинамічних систем, тобто, в тому, що вони містять гігантську кількість частинок. У цьому можна переконатися, позірно[5] розглянцвши явно необоротний процес розширення газу в пустоту на основі статистичного (молекулярно-кінетичного) методу. Отже, нехай маємо посудину, розділену перегородкою на дві однакові частини 1 і 2, причому частина 1 містить N молекул газу, а 2 – порожня. Якщо перегородку прибрати, виникне потік молекул із частини 1 у частину 2. При цьому, внаслідок хаотичності теплового руху, одразу виникне й зустрічний потік молекул з частини 2 в 1. Очевидно, що в міру зменшення кількості молекул в частині 1 і збільшення їхньої кількості в частині 2, перший потік буде з часом послаблюватись, а другий – підсилюватись. Отож через певний час, коли кількості молекул в обох посудинах стануть однаковими, ці потоки зрівняються, перетікання газу з частини 1 у частину 2 припиниться, й у системі встановиться рівноважний стан. Проте ця рівновага є динамічною – перехід молекул з однієї половини посудини в іншу не припиняється, просто кількості молекул, які перетинають у зустрічних напрямках умовну поверхню, що відділяє одну половину посудини від іншої, стають у середньому однаковими. Але в окремі моменти, знову таки через хаотичність теплового руху, вказана рівність порушується й виникає короткочасне перетікання газу з однієї частини посудини в іншу, причому байдуже, в якому напрямку. Отже, спонтанний перехід газу з другої частини в першу, тобто, обернений до розширення процес, є принципово можливим. Але йдеться тільки про дуже незначні частки молекул і про гранично малі проміжки часу. Ніхто й ніколи не спостерігав переходу з другої частини посудини в першу значної частки молекул, тим паче – всього газу. Проаналізуємо, чому? Нехай спочатку N = 1, тобто в частині 1 у початковий момент є лишень одна молекула. Після прибирання перегородки молекула буде рухатися по всій посудині, з однаковою ймовірністю перебуваючи або в другій, або в першій частині посудини. Але щоразу, коли молекула опиняється в першій половині посудини, наш “газ” спонтанно повертається в вихідний стан, причому ймовірність такого переходу дорівнює Р = 1/2, тобто, є дуже великою. Нехай тепер кількість молекул N = 2. Якщо ми будемо спостерігати за цими молекулами певний час, за який зробимо n спостережень, то виявимо, що перша молекула знаходиться в першій частині посудини в n/2 випадків. У свою чергу друга молекула в половині з цих n/2 випадків буде теж знаходиться в першій частині посудини. Тому обидві молекули будуть разом знаходитись у першій половині посудини в (1/2)(n/2) = n/4 випадків. Отже, ймовірність спонтанного повернення у вихідний стан для “газу” з двох молекул уже менша й становить Р = (1/2)·(1/2) = 1/22. Міркуючи так само, для N = 3 отримаємо Р = (1/2)·(1/2)·(1/2) = 1/23, і в загальному випадку Р = 1/2N, де N - кількість молекул у посудині. Як бачимо, спонтанний перехід газу у вихідний стан після встановлення рівноваги є принципово можливим, але ймовірність такого процесу стрімко зменшується при збільшенні кількості молекул у посудині. Так, при N = 10 імовірність Р = 1/210 = 1/1024 ≈ 0,001. Це мала, проте помітна величина: якщо “фотографувати” розподіл молекул у посудині протягом великого часу, то в середньому на кожні 1000 кадрів буде траплятись один, на якому всі молекули знаходяться в першій частині посудини. Це повністю підтверджується комп’ютерним моделюванням. Але вже при N = 100 ймовірність спонтанного повернення у вихідний стан стає Р ≈ 10-30, тобто, виявити таку подію немає жодного шансу. Реальна кількість молекул в 1 см3 газу складає ~1020 і \({P}\approx10^{-310^{19}}\). Наскільки мале це число, неможливо уявити. Тому спонтанний перехід усіх молекул газу знов у першу частину посудини абстракно є можливий, але в дісності ніколи не відбувається, і розширення газу в пустоту є необоротним. Таким чином, молекулярно-кінетична теорія не тільки пояснює причину необоротності термодинамічних процесів, а й уточнює саме поняття необоротності. А саме, необоротні процеси – це не такі процеси, що принципово не можуть самі по собі відбуватись у зворотньому напрямі, а такі, для яких сронтанний зворотній процес є гранично мало ймовірними. Відповідно, й кінцевий стан системи не є математично однозначно заданим, і
рівноважний стан є найбільш імовірним макроскопічним станом системи.
У світлі сказаного важливо з’ясувати, чим визначається ймовірність того чи іншого макроскопічного стану системи. Відповідь не важко знайти, якщо пригадати, що макроскопічний стан системи реалізується через її мікроскопічні стани, тобто, через сукупність станів окремих частинок системи (див. Вступ). Але зв'язок між мікро- та макростанами системи не є взаємно однозначним. Тоді як кожному мікроскопічному станові відповідає визначений макроскопічний стан системи, заданому макроскопічному станові відповідає безліч різних мікроскопічних станів. Наприклад, в розглянутому позірному процесі розширення газу всіх випадках (мікростанах), коли в даній частині посудини знаходиться задана кількість молекул, термодинамічні параметри (концентрація частинок, тиск, внутрішня енергія, тощо) є однаковими, тобто газ перебуває в одному й тому самому макроскопічному стані. Зважаючи на хаотичність руху молекул і величезну частоту зіткнень між ними, в МКТ прийнятий постулат про рівноймовірність усіх мікростанів, які відповідають заданому макроскопічному станові системи, тобто, – що всі мікростани трапляються однаково часто. Відтак стає зрозумілим, що заданий макроскопічний стан буде спостерігатися тим частіше, отже, тим імовірніше, чим більша кількість мікроскопічних станів йому відповідає. Тому в МКТ за міру ймовірності макроскопічного стану системи приймають його термодинамічну ймовірність або статистичну вагу Ω – кількість способів (мікростанів), якою реалізується даний макроскопічний стан системи. Наведена нижче таблиця ілюструє сказане для “газу” з чотирьох молекул 1, 2, 3, 4. У ній показані всі можливі розподіли молекул по частинах посудини (мікростани), відповідні макростани та їхні термодинамічна Ω й математична Р ймовірності. Як видно, рівноважний стан, у якому молекули розподілені по посудині рівномірно, є найбільш імовірним (Ω = 6), а стани, в яких усі молекули самодовільно скупчуються тільки в частині 1, або тільки в частині 2, є найменш імовірними (Ω = 1).
| Способи реалізації макростанів | Ω | P | |
| A | B | ||
|
0 |
1, 2, 3, 4 | 1 | 1/16 |
|
1 2 3 4 |
2, 3, 4 1, 3, 4 1, 2, 4 1, 2, 3 |
4 | 4/16 |
|
1, 2 1, 3 1, 4 2, 3 2, 4 3, 4 |
3, 4 2, 4 2, 3 1, 4 1, 3 1, 2 |
6 | 6/16 |
|
1, 2, 3 1, 2, 4 1, 3, 4 2, 3, 4 |
4 3 2 1 |
4 | 4/16 |
| 1, 2, 3, 4 | 0 | 1 | 1/16 |
| Усього мікростанів 24 = 16 | |||
Поняття термодинамічної ймовірності дозволяє кількісно виразити зв’язок між ентропією та ступенем наближеності системи до термодинамічної рівноваги, про який говорилося вище. Це зробив Л. Больцман, який довів, що ентропія S системи в стані із термодинамічною ймовірністю Ω визначається, як
|
\( {S}=k\ln\Omega\). |
(8.17) |
де k = 1,38·10-23 Дж/К – вже відома нам з МКТ “стала Больцмана”.
Вираз (8.17), який називається формулою Больцмана, є статистичним означенням поняття ентропії. Формула Больцмана відіграє велику роль у статистичній фізиці та термодинаміці. Зокрема, вона розкриває глибинний зміст закону зростання ентропії. А саме, цей закон указує на те, що всі процеси в природі йдуть у напрямку зростання молекулярного безпорядку в системі. Це в свій час спонукало дискусію про так звану “теплову смерть Всесвіту”, тобто, про неминучий перехід нашого Всесвіту до стану термодинамічної рівноваги й припинення в ньому будь-яких процесів. Це, в принципі, не виключено, але сьогодні для однозначної відповіді на це питання наявні знання про походження та еволюцію Всесвіту є недостатніми.
2. Яка фізична величина називається ентропією? В яких одиницях вона вимірюється? Які має основні властивості?
3. Чому не можна безпосередньо визначити зміну ентропії в заданому необоротному процесі?
4. Чи можна взагалі визначити зміну ентропії в необоротному процесі? Чому? Як?
5. У чому полягає закон зростання ентропії? Чи може ентропія термодинамічної системи зменшуватись? За яких умов?
6. Дайте означення рівноважного стану системи на основі поняття ентропії.
7. Який статистичний зміст має ентропія?
8. Що таке “термодинамічна імовірність” макроскопічного стану системи. Який зв’язок існує між нею та ентропією?
[1] Див. «Фізика для бакалаврів. Механіка», розділ IV, п.2.2.
[2] Див. розділ V, п. 3.
[3] Нагадаємо, що в необоротному процесі параметри стану не мають визначених значень для всієї системи, тому процес неможливо зобразити якоюсь лінією на графіку.
[4] Конкретним прикладом цього є результат наведеного в п. 2 розрахунку зміни ентропії при розширенні газу в пустоту.
[5] Це означає “удавано” і є одним із способів доведення. До прикладу, у фізиці нерідко розглядають “позірні експерименти” – удавані досліди, які не суперечить фізичним законам, але технічно є неможливими.