ФІЗИКА ДЛЯ БАКАЛАВРІВ. МОЛЕКУЛЯРНА ФІЗИКА ТА ТЕРМОДИНАМІКА
ІІ. ВЛАСТИВОСТІ ГАЗІВ
У газах сили притягання між молекулами малі, тому молекули не утримуються одна біля одної, а рухаються по всьому наданому об’єму. При не дуже низьких температурах середня кінетична енергія теплового руху молекул у газі істотно перевищує енергію зчеплення між ними. За таких умов можна вважати, що молекули в газах рухаються як вільні частинки, взаємодіючи лише при безпосередніх зіткненнях між собою та зі стінками посудини.
З іншого боку, при не дуже високих тисках концентрація молекул у газах є порівняно невелика, через що власний об’єм молекул виявляється набагато меншим, ніж об’єм посудини, в якій знаходиться газ. Наприклад, при кімнатній температурі й нормальному атмосферному тискові об’єм молекул складає ≈ 0,1% об’єму посудини, і на практиці власні розміри молекул можна не враховувати. Тому, розглядаючи гази, широко використовують спрощену модель “ідеальний газ” - систему, що складається з частинок несуттєвих розмірів, які взаємодіють лише при випадкових пружних зіткненнях між собою та зі стінками посудини. Інакше говорячи молекули ідеального газу не мають власного об’єму та потенціальної енергії взаємодії між собою[1]. Така модель добре відображає властивості справжніх газів за звичайних умов. При екстремальних умовах (наднизькі температури та надвисокі тиски) наявність власного об’єму та взаємодії між молекулами даються взнаки і властивості реальних газів стають більш складними. Але властивості ідеального газу можна теоретично встановити на основі лише простих молекулярно-кінетичних уявлень.
Нижче розглянуті наступні питання
2.1. Тиск ідеального газу
2.2. Рівняння Клапейрона. Ізопроцеси в газах
2.3. Реальний газ
|
|
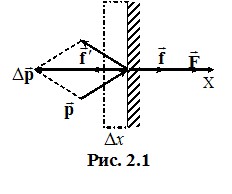
Газ створює тиск на стінки посудини завдяки ударам молекул, причому в ідеальному газі ці удари є абсолютно пружними. Розглянемо дію молекул газу на малу ділянку стінки посудини, рис. 2.1.
Під час зіткнення на молекулу з боку стінки діє мікроскопічна сила, середня величина якої дорівнює відношенню зміни імпульсу молекули до тривалості зіткнення (другий закон Ньютона): \(\vec{f}'=\Delta{\vec{p}}/\tau \). За третім законом Ньютона з боку молекули на стінку діє сила \(\vec{f}=-\vec{f}' \). Можна сказати, що ця сила дорівнює імпульсу, переданому молекулою стінці за одиницю часу. Величина Δp є дуже малою, тому дія молекул газу на стінку посудини має характер мікроскопічних миттєвих “уколів”, урахувати які безпосередньо неможливо. Але у тому й немає потреби - через макроскопічну кількість молекул зіткнення відбуваються так часто та щільно, що створюють рівномірно розподілену по поверхні стінки сталу силу тиску F. Ця сила дорівнює сумарному імпульсу, що передається стінці всіма молекулами, котрі стикаються з нею протягом 1 с. Величину F можна визначити за допомогою таких міркувань. Пружне зіткнення молекули зі стінкою відбувається без утрати кінетичної енергії та величини швидкості. Тому молекула відлітає від стінки з таким самим за модулем імпульсу й під таким самим кутом, як і налітає. При цьому модуль переданого стінці імпульсу складає (див. рис. 2.1):
\(\left|\Delta{\vec{p}}\right|=2p\cos\alpha=2mv\cos\alpha \) \( \Rightarrow \) \( \left|\Delta{\vec{p}}\right|={2mv_x}\),
де m - маса молекули, α – кут між вектором імпульсу молекули та нормаллю до стінки, vx - проекція на вісь Х швидкості, з якою молекула налітає на стінку. Для визначення сили тиску припустимо спочатку, що всі молекули, котрі налітають на стінку, мають однакову величину vx. Тоді сила тиску
\({F}=\nu\cdot{2mv_x}\).
де \(\nu \) - кількість зіткнень молекул зі стінкою за 1 с. Щоб знайти ν, виділимо біля стінки шар молекул настільки малої товщини Δx, що в його межах молекули не стикаються між собою. Тоді всі молекули в шарі, що рухаються до стінки, зіткнуться з нею протягом часу Δt = Δx/vx. Кількість таких молекул ΔN′ дорівнює половині загальної кількості молекул у шарі. Тоді, якщо концентрація молекул газу (кількість в одиниці об’єму) дорівнює n і площа ділянки стінки S, то ΔN′ = nSΔx/2, і кількість зіткнень за одиницю часу
\(\nu=\frac{\Delta{N'}}{\Delta{t}}=\frac{1}{2}nx_xS \) і \({F=nmv_v^2 S}\).
Насправді молекули мають різні швидкості, тому реальна сила тиску визначається середнім значенням квадрата проекції швидкості \(\left\langle{v_x^2}\right\rangle \):
|
\({F}=nm\left\langle{v_x^2}\right\rangle{S}\). |
(2.1) |
Усі напрямки руху молекул є рівноймовірними й незалежними, тому можна записати
\(\left\langle{v_x^2}\right\rangle=\left\langle{v_y^2}\right\rangle=\left\langle{v_z^2}\right\rangle \) і \(\left\langle{v^2}\right\rangle=\left\langle{v_x^2}\right\rangle+\left\langle{v_y^2}\right\rangle+\left\langle{v_z^2}\right\rangle=3\left\langle{v_x^2}\right\rangle \) \(\Rightarrow \) \(\left\langle{v_x^2}\right\rangle=\frac{1}{3}\left\langle{v^2}\right\rangle \). .
Підставивши цей вираз у (2.1) і поділивши на площу S, отримуємо для тиску ідеального газу:
|
\({P}=\frac{1}{3}nm\left\langle{v^2}\right\rangle \) |
(2.2) |
або
|
\({P}=\frac{2}{3}n\left\langle{E}\right\rangle\). |
(2.2а) |
Ці рівняння є принципово важливими, оскільки вони встановлюють теоретичний зв’язок між макроскопічним параметром газу - тиском і мікроскопічними параметрами - масою та швидкістю теплового руху молекул, або їх кінетичною енергією. Тому рівняння (2.2) або (2.2а) часто називають основним рівнянням молекулярно-кінетичної теорії газу. З них випливають важливі наслідки.
Перш за все зауважимо, що в (2.2а) фігурує середня енергія поступального руху молекул, яка визначається тільки температурою газу, згідно з (1.2). Тому для тиску можна записати:
|
\({P=nkT}\). |
(2.3) |
Отже, тиск ідеального газу не залежить від його природи й визначається тільки концентрацією молекул та температурою газу. Зокрема, це означає, що при однакових температурі та тиску всі гази мають однакову концентрацію молекул (закон Авогадро). За нормальних умов (температура 0 оС (273 К) і тиск 760 мм. рт. ст (1,01·105 Па)) вона складає п0 = 2,69·1025 1/м3 і називається числом Лошмідта. При цьому 1 моль будь-якого газу за нормальних умов займає однаковий об’єм V0 = 22,4·10-3 м3. Із незалежності тиску газу від природи молекул випливає, що вираз (2.3) є чинним не лише для однорідних газів, а й для суміші газів, які не вступають між собою в хімічні реакції. У цьому випадку
\( {n}=n_{1}+n_{2}+...+n_{i}+...=\sum_{i}n_{i}\),
де ni - концентрації компонентів суміші. Відповідно, тиск суміші
|
\( {P}=\left(\sum_{i}n_{i}\right){kT}=\sum_{i}(n_{i}kT) \) \( \Rightarrow \) \( {P}=\sum_{i}P_{i}\), |
(2.4) |
де \({P_i}=n_ikT \) - парціальні тиски компонентів, тобто тиски, які створювала би в посудині дана компонента суміші за відсутності всіх інших.
|
\({R=N_Ak}\) = 8,31 Дж/(моль·К) |
(2.5) |
називається універсальною газовою сталою. Врахувавши це, отримуємо рівняння стану ідеального газу, яке називається рівнянням Клапейрона (Клапейрона-Мендєлєєва):
|
\({PV=\nu{RT}=\frac{m}{M}RT}\), |
(2.6) |
де \({\nu}\) - кількість молів, т - маса і М - молярна маса газу. Для одного моля газу.
\({PV=RT}\).
Ізопроцеси в газах. Рівняння Клапейрона (2.6) дозволяє аналізувати не лише рівноважні стани, а й рівноважні процеси в газах, тобто з’ясовувати характер зміни термодинамічних параметрів газу за тих чи інших умов. Для теорії найбільший інтерес викликають так звані ізопроцеси, тобто, процеси, що відбуваються з незмінною кількістю газу і при незмінному значенні одного з параметрів стану. Розрізняють ізотермічний (\(\nu=\mathrm{const}\), T = const), ізобарний (\(\nu=\mathrm{const}\), Р = const) та ізохорний (\(\nu=\mathrm{const}\), V = const) процеси.
В ізотермічному процесі права частина (2.6) не змінюється, отже
|
PV = const, де const = νRT. |
(2.7) |
Це рівняння виражає закон Бойля-Маріотта:
при постійній температурі добуток тиску заданої кількості газу на об’єм є сталим .
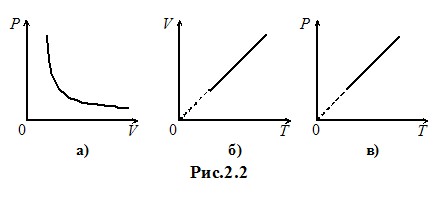
Графік, що показує зв’язок між параметрами стану в ізотермічному процесі, називається ізотермою і на діаграмі (Р, V) для ідеального газу має форму гіперболи (рис. 2.2а).
|
|
Ізобарний (ізобаричний) процес відбувається при сталому тиску газу, тому на основі (2.6) можна записати:
|
\({V=aT}\), де \({a}=\frac{mR}{MP}=\mathrm{const}\). |
(2.8) |
Це є вираз закоу Гей-Люссака:
при сталому тиску об’єм заданої кількості газу змінюється прямо пропорційно абсолютній температурі.
Графік цього процесу (рис. 2.2б називається ізобарою. на діаграмі (V, Т).
В ізохорному (ізохоричному) процесі подібним чином змінюється тиск:
|
\({P=bT}\), де \({b=}\frac{mR}{MV}=\mathrm{const}\). |
(2.9) |
Це є аналітичний вираз закону Шарля:
тиск заданої кількості газу при сталому об’ємі є прямо пропорційним температурі. Вигляд ізохори на діаграмі (P, T) показано на рис. 2.2в.
Рівняння (2.8) і (2.9) інколи записують через температуру в шкалі Цельсія, роблячи в (2.8) і (2.9) заміну Т = 273 + t. Тоді
|
\({V=V_0(1+\alpha{t})}\) і \({P=P_o(1+\beta{t})}\), |
(2.10) |
де V0 = 273 та Р0 = 273 – об’єм і тиск при 0 °С (273 К); α - термічний коефіцієнт об’єму (коефіцієнт об’ємного розширення) і β - термічний коефіцієнт тиску. Очевидно, що
\(\alpha=\beta=\frac{1}{273}\,\,K^{-1}\).
Рівняння Ван дер-Ваальса. Найпроостішим і зручним для якісного аналізу властивостей реальних газів є рівняння стану, що запропонував Ван-дер-Ваальс на основі досить простих міркувань. Об’єм V, який фігурує в рівняннях (2.6), (2.6а) є об’ємом посудини, в якій міститься газ. В дійсності через наявність власних розмірів для молекул доступний не весь об’єм посудини. Тому в рівнянні стану замість V має фігурувати величина V - b, де b - поправка на власний об’єм молекул, яка приблизно дорівнює добутку об’єму однієї молекули на їх кількість[3]. Відповідно, тиск 1 моля реального газу мав би виражатися, як
|
\({P}=\frac{RT}{V_0-b}\), |
(2.11) |
|
|
|
де V0 - молярний об’єм газу (об’єм 1 моля).
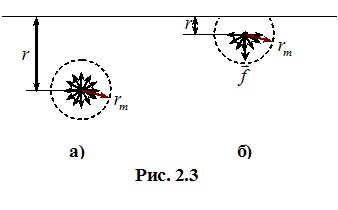
Але на тиск газу впливають і сили притягання між молекулами. Для аналізу цього впливу вводять поняття радіуса молекулярної дії rm, тобто максимальної відстані між молекулами, на якій сили притягання ще мають помітну величину (ця відстань є досить умовною і складає ~10-9 м, тобто декілька поперечників молекули). Якщо молекула знаходиться на відстані r > rm від стінки посудини, то вона оточена сусідніми молекулами з усіх боків у середньому рівномірно і їхня дія є компенсованою (рис.2.3а). Але біля стінки, коли r < rm, кількість сусідніх молекул, які тягнуть дану молекулу донизу, переважає кількість тих, які тягнуть її вгору. Тому на кожну молекулу біля стінки з боку інших діє певна результуюча сила \(\vec{f}\) , спрямована всередину газу (рис. 2.3б). Такі молекулярні сили сповільнюють рух молекул на підльоті до стінки і, згідно з (2.2), зменшують тиск газу на стінку на деяку величину Pi. З урахуванням цього, рівняння (7.13) слід записувати у вигляді
|
\({P}=\frac{RT}{V_0-b}-P_i \). |
(2.11а) |
Величина \({P_i}\) визначається сумарною силою, що діє на молекули в прилеглому до стінки посудини шарі газу. Ця сила є пропорційна як кількості молекул у пристіночному шарі, на які діють молекулярні сили, так і кількості молекул у сусідньому шарі, що створюють її. Обидва шари мають приблизно однакову товщину порядку rm і, відповідно, однакову кількість молекул, прямо пропорційну концентрації газу n. Тому Pi ~ n2 . У свою чергу концентрація молекул є обернено пропорційна об’єму газу (для 1 моля n = NA/V0), отже можна записати \({P_i}=a/V_0^2 \), де а - деяка стала. У підсумку, рівняння (2.11а) набуває вигляду
\({P}=\frac{RT}{V_0-b}-\frac{a}{V_0^2}\),
або
|
\(\left(P+\frac{a}{V_0^2}\right)\left(V_0-b\right)={RT}\). |
(2.12) |
Для \(\nu \) молів газу \({V=\nu{V_0}}\), відповідно, рівняння (2.12) набуває вигляду:
|
\(\left(P+\frac{a\nu^2}{V^2}\right)\left(V-\nu{b}\right)=\nu{RT}\). |
(2.12а) |
Рівняння стану реального газу (2.12) або (2.12 а) називається рівнянням Ван-дер-Ваальса.
Ізотерми Ван-дер-Ваальса. Критичний стан. Подивимось, які специфічні властивості реальних газів передрікає рівняння Ван-дер-Ваальса та як теоретичні прогнози узгоджуються з дослідними фактами. Для цього проаналізуємо ізотерми Ван-дер-Ваальса, тобто залежності тиску газу від об’єму при сталих температурах, які випливають із рівнянь (2.12), (2.12 а). На рис. 2.4 показаний характерний вигляд теоретичних ізотерм Ван-дер-Ваальса для різних температур газу. При високих температурах, як і слід очікувати, вони нагадують ізотерми ідеального газу - при розширенні тиск монотонно зменшується (крива 1). Але при низьких температурах на кривих з’являється характерна хвиляста ділянка (крива 3).
|
|
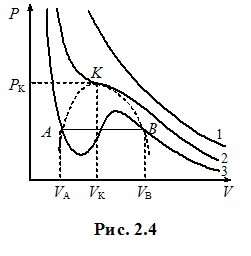
Ці два різні типи залежностей розділяються так званою критичною ізотермою (крива 2), яка відповідає певній критичній температурі Тк і має точку перегину К, яка називається критичною точкою. Цій точці, крім критичної температури Тк, відповідає критичнйм тиск Рк і критичний об’єм Vк. Критичні параметри можна знайти, виходячи з того, що в точці К (точці перегину) перша та друга похідні тиску по об’єму дорівнюють нулю. Відповідні розрахунки на основі (2.12) для одного моля газу дають:
|
\( {V}_{0k}=3b \), \( {P}_{0k}=\frac{a}{27b^{2}}\), \( {T}_k=\frac{8a}{27Rb}\) |
(2.13) |
Як видно, критичні параметри визначаються виключно поправками Ван-дер-Ваальса а і b, тобто індивідуальними властивостями молекул. Але величина Кк = RTk/P0kV0k, яка називається критичним коефіцієнтом, є універсальною: згідно з (2.13) її значення, не≈залежно ні від чого, складає Кк = 8/3 ≈ 2,67. Дослід свідчить, що це не зовсім так: критичний коефіцієнт є дещо більшим і залежним від газу[4]. Отже, рівняння Ван-дер-Ваальса не є точним. Про це свідчить і наявність підйомів на хвилястих ділянках ізотерм при T < Tk (крива 3, рис. 2.4), які відповідають зростанню тиску при розширенні газу. Це є неможливо, бо означає збільшення тиску при зменшенні концентрації молекул при незмінній температурі. І дійсно, в експерименті указаний аномальний підйом на ізотермах реального газу не спостерігається. Але натомість виникає “поличка” (відрізок АВ на кривій 3, рис. 2.4): при зменшенні об’єму від VВ до VА тиск газу залишається сталим, а потім стрімко зростає. Спостереження показують, що це пов’язано з конденсацією газу в рідину. У процесі стискання при об’ємі VВ в посудині крім газу з’являється рідина, якої дедалі стає більше, а газу - менше. При об’ємі VА конденсація закінчується, і в посудині міститься лише рідина. Цим пояснюється й дуже стрімке зростання тиску при подальшому стисненні – адже рідина є практично нестисливою.
Таким чином, рівняння Ван-дер-Ваальса показує можливість переходу газу в рідину при ізотермічному стисканні. При цьому об’єм речовини під поршнем без зміни тиску зменшується від значення VА, яка відповідає початковому об’єму газу, до VВ – об’єму рідини, що утворилася з усього газу. Інтервал значень об’єму VВ - VА, при якому в посудині співіснують газоподібна та рідка фази речовини, що показаний штриховою кривою на рис. 2.4, із збільшенням температури зменшується і при Т = Тк зникає зовсім. При цьому, як показує дослід, при V > Vk у посудині спостерігається лише газ, а при V < Vk - лише рідина. Отже, при переході через критичну точку К увесь газ одномоментно перетворюється на рідину, проминаючи двофазну стадію. Самій точці К(Рk, Vk , Тk) відповідає специфічний критичний стан речовини, при якому зникає будь-яка відміна між газом і рідиною. Зокрема, зникає меніск – межа поділу між рідиною та газом і спостерігається опалесценція - різке помутніння речовини по всьому об’єму безпосередньо перед переходом у рідкий стан. При температурах Т > Тк ізотерми Ван-дер-Ваальса не мають розглянутих особливостей, і можна припустити, що за таких умов газ весь час лишається газом, тобто його ніяким способом неможливо перевести в рідкий стан. Це дійсно так: практика отримання скраплених газів свідчить, що будь-який газ можна стисканням перетворити на рідину тільки при температурі, нижче критичної.
Підводячи підсумок, відмітимо, що рівняння Ван-дер-Ваальса якісно правильно відображає властивості реальних газів у широкому діапазоні параметрів стану. І це гідне подиву, оскільки в цьому рівнянні враховані не детальні властивості сил взаємодії між молекулами, а, за великим рахунком, лише сам факт існування таких сил.
1. Що таке“ідеальний газ”? За яких умов природні гази можна розглядати як ідеальні?
2. Чому при високих тисках і низьких температурах властивості природних газів відрізняються від ідеальних?
3. Чим тиск газу на стінки посудини відрізняється від тиску рідини?
4. Якими мікроскопічними параметрами визначається тиск газу на стінки посудини?
5. Якими макроскопічними параметрами визначається тиск газу на стінки посудини?
6. Як визначається тиск суміші газів?
7. Що називається рівнянням стану та який вигляд воно має для ідеальних газів?
8. Чому рівняння Клапейрона можна використовувати для аналізу рівноважних процесів у газі і не можна – для нерівноважних?
9. Що таке ізопроцеси? Перелічіть ізопроцеси в газах.
10. Як виглядають ізотерми на діаграмі (Р, Т)? (V,T)?
11. Як виглядають ізобари на діаграмі (Р, V)? (P,T)?
12. Як виглядають ізохори на діаграмі (Р, V)? (V,T)?
13. Як впливає на тиск реального газу (порівняно з ідеальним) наявність розмірів у молекул? Як це пояснити, виходячи з механізму створення тиску?
14. Як впливає на тиск реального газу (порівняно з ідеальним) наявність сил притягання між молекулами? Як це пояснити, виходячи з механізму створення тиску?
15. Поясніть, які процеси в реальному газі відображають різні характерні ділянки ізотерм Ван-дер-Ваальса.
16. За яких умов газ при стисканні скраплюється (переходить у рідкий стан), а за яких – ні.
17. Що таке критичний стан речовини? Чи можна його спостерігати в експерименті?
[1] Але в деяких випадках доводиться враховувати рух і взаємодію атомів у молекулах.
[2] Атмосферний тиск вимірюють у позасистемних одиницях – міліметрах ртутного стовпчика (1 мм рт. ст. = 133,3 Па).
[3] Насправді вона дещо більша, оскільки навіть при щільній упаковці між молекулами лишаються певні не заповнені проміжки.